ModBase
ModBase Documentation
ModBase is a comprehensive database of comparative protein structure models.
ModBase is organized into datasets, which are either available to the public, to the academic community, or to specific users.
To submit new comments/suggestions/bugs, please send email to .
| Help Topics | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
|
ModBase Search | ||||||||||||
|
User Login There are several kinds of possible user logins that can be managed at the dataset management page:
For a ModWeb dataset, the user receives username/password by email after
the modweb calculation has been finished and the data have been stored in ModBase. | ||||||||||||
|
Datasets
ModBase is organized into Datasets. The comprehensive Dataset (combination of all SP/TR datasets) comprises comparative models of
all sequences in the SwissProt and TrEMBL databases that have detectable similarity to an experimental protein
structure. | ||||||||||||
|
Dataset Selection To select a specific subset of the datasets available to you, please go to the search page, and click on "Select specific dataset(s)". This will open two boxes. The left box contains all datasets available to you. The right box contains a subset of those. You can either click on a dataset on the left, and then click the arrow, or double click a dataset on the left, to include it into the search. You can also double click a dataset on the right to exclude it from the search. If you don't use this menu (Collapse dataset selection), all available datasets are selected by default. | ||||||||||||
|
Search Types Different search options are available in ModBase.
| ||||||||||||
|
Display Types Depending on the chosen search mode, different display options are available in ModBase.
| ||||||||||||
|
Search Properties Search properties are dependent on the chosen search mode. The most common are described here:
| ||||||||||||
|
Advanced Search Properties
Search options for model properties are available: | ||||||||||||
|
Original Sequence ModPipe, the software that calculates the ModBase models, modifies the original protein sequences to replace non-standard amino acid residues. The Original Sequence represents the un-modified version, gets only displayed if a modification has taken place. | ||||||||||||
|
Input Sequence Enter a sequences in either FASTA format or just the amino acid residues. Non-standard residues are ignored. | ||||||||||||
|
FASTA Format
| ||||||||||||
|
Sequence Similarity Search Search by sequence similarity using BLAST. | ||||||||||||
|
Action Pulldown Menus The Action Pulldown Menus give the users the option to:
| ||||||||||||
|
Linking to ModBase Models from external databases
To link from outside pages to specific ModBase sequences/models, please use the following link construction:
| ||||||||||||
|
Model Details page This is the default ModBase page for models for one sequence. Sequence information, model/sequence coverage and model information are displayed. Two version of this page are available: Graphical and schematic | ||||||||||||
|
Model Details (Graphical) The graphical Model Details page gives access to all available information for the models of one sequence: Sequence information, model information, database crosslinks. If there are several models for the current sequence, the model with the highest sequence identity to its template is displayed, and thumbprints of the other models are show as well. Mouseover the thumbprint to get information about that model. | ||||||||||||
|
Model Coverage Sketch
If (a) specific dataset(s) have been selected, and the query sequence has been modeled in other available datasets
as well, the darker colors indicate the selected dataset(s) and the light colors the others.
| ||||||||||||
|
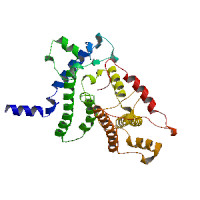
Model Image The model images are created on the fly using MolScript and raster 3d. | ||||||||||||
|
Model Details (Schema) The schematic model details page shows a thumbprint of each model and a sketch of the model/sequence coverage. | ||||||||||||
|
Filtered Models
ModBase contains many models for some sequences. This might be due to a very long sequence, or because this
sequence has been processed in several datasets. To avoid confusion, a "filtered" subset of the models is
displayed on the model details pages, containing only the
models from the last calculation.
Please click on "all models" if want to check out all available models, a better one might be hiding.
On the schematic page, a mouseover the
thumbprint gives more information about that model. | ||||||||||||
|
Sequence/Model Overview This is the default page when the search results in more than one sequences. There are two models: Sequence Overview and Model Overview. | ||||||||||||
|
Sequence Overview The Sequence Overview page summarizes the search results for many sequences. The sequence coverage Sketch indicates the modeled area(s) for the given sequence. Click on the sketch to go to the "Model Details" page. | ||||||||||||
|
Model Coverage Sketch Similar as the Model Details Sketch, but smaller and with less complexity. | ||||||||||||
|
Model Overview The Model Overview page displays the search results as one line for each model. Details about modeling quality and templates are being displayed. Click on the thumbprint on the left to get to the "Model Details" page for that model. | ||||||||||||
Model/Fold Reliability
Please click on the Ball to go to the Model Details (schema) page for this model/sequence. | ||||||||||||
|
Model Thumbnail Please click on the Thumbnail to go the the Model Details (graphical) page for this model. | ||||||||||||
|
Sequence Information | ||||||||||||
|
Primary Sequence Database ID
The Sequence Database ID displayed on the "Sequence Information" section is chosen according to the following
order of availability: | ||||||||||||
|
Original Sequence Database ID The original Sequence Database ID from the fasta file that was used for the modeling calculation. The prefix "CU" indicates a custom database ID. This ID can be useful to identity sequences from modweb calculations. | ||||||||||||
|
Sequence Length Length of the input sequence. AnnotationThe Sequence Annotation is either retrieved from UniProt or GenPept, or, if those are not available, from the input fasta file. Organism Information (Taxonomy)
ModBase currently contains a number of datasets of complete genomes. These are included in the pull-down menu. | ||||||||||||
|
Model Information ModBase often has several models for one sequence. This can happen, if the sequence got processed in different datasets (at different times, for a different project or with different hit-selection criteria), or if there are models for different domains, because the template PDB structures don't cover the full target sequence. Also, ModPipe usually calculates a large number of models for each sequence section, and use a number of quality criteria to select the "best". Since the quality criteria are not always in agreement, ModPipe frequently chooses up to four models for each region | ||||||||||||
|
Alignment Significance Significance of the alignment between the target and the template as reported by NCBI's PSI-BLAST program (Nucl. Acids Res. 25, 3389-3402, 1997). This is the significance reported during the template (PDB) database search. It is not the significance of the modeling alignment produced by Modeller. | ||||||||||||
|
E-Value ModPipe1.0: Significance of the alignment between the target and the template as reported by NCBI's PSI-BLAST program (Nucl. Acids Res. 25, 3389-3402, 1997). This is the significance reported during the template (PDB) database search. It is not the significance of the modeling alignment produced by Modeller. ModPipe2 and later:Similar significance value, but calculated by Modeller using the Build-Profile routine. | ||||||||||||
|
GA341 (Model Score) Score for the reliability of a Model, derived from statistical potentials (F. Melo, R. Sanchez, A. Sali,2002 PDF). A model is predicted to be good when the model score is higher than a pre-specified cutoff (0.7). A reliable model has a probability of the correct fold that is larger than 95%. A fold is correct when at least 30% of its Cα atoms superpose within 3.5 Å of their correct positions. | ||||||||||||
|
Protein Size Length of the modeled sequences (original sequence, not the modeled part). | ||||||||||||
|
Model Size Length of the model; | ||||||||||||
|
Reliable Model / Fold Assignment A model is considered to be reliable (have a reliable fold assignment) if it is evaluated within the following thresholds by at least one of theses model evaluation criteria:
| ||||||||||||
|
Sequence Identity Percentage of identical residues in the alignment between the target and the template as reported during the template search. | ||||||||||||
|
ModPipe Protein Quality Score The ModPipe Protein Quality Score is a composite score comprising sequence identity to the template, coverage, and the three individual scores evalue, z-Dope and GA341. We consider a MPQS of >1.1 as reliable. | ||||||||||||
TSVMod
Reference: D. Eramian, N. Eswar, M.Y. Shen, A. Sali. How well can the accuracy of comparative protein structure models be predicted? Protein Sci 17, 1881-1893, 2008. | ||||||||||||
|
z-Dope Using the probability theory, we derive an atomic distance- dependent statistical potential from a sample of native structures that does not depend on any adjustable parameters (Discrete Optimized Protein Energy, or DOPE). DOPE is based on an improved reference state that corresponds to noninteracting atoms in a homogeneous sphere with the radius dependent on a sample native structure; it thus accounts for the finite and spherical shape of the native structures. | ||||||||||||
|
Target Region The region of the protein sequence that is modeled. | ||||||||||||
|
Protein Length The length of the original protein sequence. | ||||||||||||
|
Template PDB Code The PDB code of the template the model is based on. | ||||||||||||
|
Template Region The region of the PDB structure that was used as a template. | ||||||||||||
|
ModPipe Version ModPipe is the underlying software pipeline that is used to build all ModBase models. ModPipe1.0 relies on PSI-Blast and Impala for template selection and fold assignment. ModPipe2 is additionally using the Build-Profile method in Modeller. ModPipe2 models are also scored with the MPQS and z-Dope. | ||||||||||||
|
ModPipe Date Modeling date for current model. Often, a sequence got modeled in several independed datasets. If you model is older, please check the additional models (by clicking on the thumbprints below the prominent model) for a newer date. If you suspect that a better template has been released after the newest model date, you should submit the sequence to ModWeb to get a current model. | ||||||||||||
|
Coordinate (3D) File Coordinate file for the model in the PDB format. The "fifth column" (which normally contains B-factors or order parameters) contains the Modeller error profile. | ||||||||||||
|
Modeller Error Profile
| ||||||||||||
|
PAP Alignment Format The 'PAP' format is nicer to look at than the 'PIR' format, but not as computer friendly. The alignment.write() command description in the Modeller manual contains more detailed information about this format. | ||||||||||||
|
PIR Alignment Format The 'PIR' format resembles that of the PIR sequence database. It is described in the Modeller manual and is used for comparative modeling with Modeller because it can contain all the information useful for modeling. | ||||||||||||
|
LigBase (as integrated in ModBase)
LigBase is a structural database of ligand binding sites. The LigBase database tables
contain all amino acid residues that are within 5 Angstroms from a small molecule ligand in a given PDB file.
The current version of LigBase contains ligand binding information from 16629 PDB files. 1. Putative ligand binding sites derived from the template
Putative ligand binding sites of ModBase models are derived from the template on the fly by
parsing the ModBase alignment file. The native ligand binding residues of the template (TEMPL) and the
derived ligand binding sites of the model (MODEL) are shown. Additionally, the putative binding residues of
the model are colored in its image. If a gap is found in the alignment at a ligand binding residue, a "-"
is displayed instead of the model residue.
Many PDB files used as templates don't include a ligand, but closely related PDB files might have one or more ligands bound. Using DBALI , a database of structural alignments, and using the information in the PDB-LigBase tables, the INHERITOR table of LigBase contains the amino acid residues of PDB files with ligands and the equivalent residues of related PDB files. Additionally, the sequence identity and coverage between those PDB files and between the respective binding sites are stored. Once the INHERITOR information is retrieved, putative ligand binding sites from related PDB files are determined similar to the binding sites derived from the templates. The inherited residues are shown (INHER) together with the equivalent model residues (MODEL). LigBase model coverage sketchThe ligbase coverage sketch displays the same information as the general model coverage sketch. Additionally, it displays the position of the amino acid residues which are putative ligand binding residues. | ||||||||||||
|
ABC Transporter Datasets The ABC Transporter model dataset includes domains in all 48 human ABC transporters. It also includes models of disease-associated and polymorphic non-synonymous SNPs found in the nucleotide binding domains. | ||||||||||||
|
LS-Mut LS-Mut is a database of Somatic mutations found in advanced pancreatic tumor or glioblastoma multiforme from the Karchin lab at Johns Hopkins University. Please refer to the following publications:
| ||||||||||||
|
ModWeb
When a ModWeb job is finished (with the option of depositing the models into ModBase), the user gets a results page (and optionally an email)
including the dataset name, and the username/password for ModBase to access that particular dataset.
If you still have problems, please email . | ||||||||||||
|
ModBase Command line Retrieval
| ||||||||||||
|
Predicted Protein Complexes MODBASE contains structure-based predictions of 3,213 binary and 1,234 higher order protein complexes in Saccharomyces cerevisiae involving 750 and 195 proteins, respectively. To generte candidate complexes, comparative models of individual proteins were built and combined together using complexes of known structure as templates. These candidate complexes were then assessed using a statistical potential, derived from binary domain interfaces in PIBASE (https://salilab.org/pibase). A benchmark indicates a false positive rate of 3% and a true positive rate of 97%. Moreover, the predicted complexes are also filtered using functional annotation (http://yeastgenome.org) and sub-cellular localization (https://yeastgfp.yeastgenome.org/) data. | ||||||||||||
SNP Stability
| ||||||||||||
|
Modeling Leverage Calculations The modeling leverage of a PDB structure is calculated by:
All resulting models are deposited in ModBase. | ||||||||||||





{kind=link}