 MultiFit Webserver Help
MultiFit Webserver Help
MultiFit Help Pages
Input Fields
Email
If set, a notification will be sent once the job is completed.
Complex density map
The assembly density map in MRC format.
Resolution
The resolution of the assembly density map in angstroms.
Voxel spacing
The dimension of a single voxel.
Contour level
Consider voxels with density values above the threshold.
X, Y, and Z origins
Optional parameters for x, y, and z origins. If not given, origins are set to 0, 0, 0.
symmetry mode
Select if the complex is cyclic symmetric or non symmetric. Symmetry is used in the sampling procedure for cyclic symmetric structures.
Symmetry order
The symmetry order of the ring (3 for trimer, 4 for tetramer etc.).
Subunit coordinate file
Upload coordinate file of the subunit in PDB format.
Reset button
Reset all the input fields.
Output
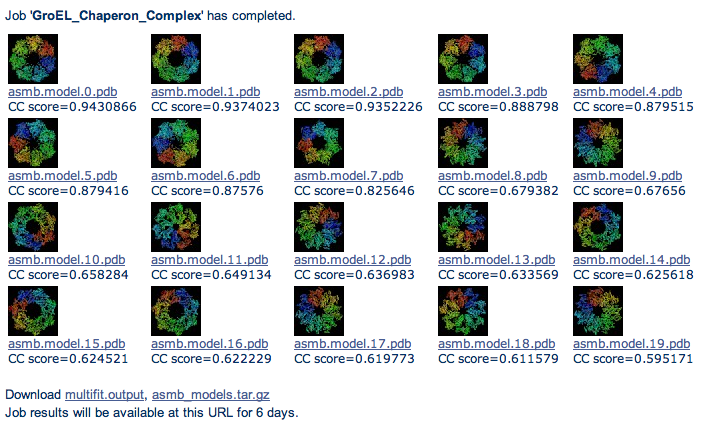
The output is a set of models that best fit the density map.
The first top 20 models are displayed as a table ranked from top-left to bottom-right and the complete list
of models is found in the transformations output file (multifit.output).
Top 20 models
MultiFit.output file
This output file reports MultiFit assembly modeling solutions. Each line of 11 fields defined a single model. The solutions are sorted by fitting scores.Solution index
The index of the solution.
Solution filename
Filename of the solution
Fit rotation
The rotation part (in quaternions) of a transformation found by a fiiting procedure.
Fit translation
The translation part of a transformation found by a fiiting procedure.
Match size
If anchor points matching is used, this field stores the size of the match.
Match average distance
If anchor points matching is used, this field stores the RMSD between the model anchor points and the density anchor points.
Match cluster size
If anchor points matching is used, this field stores the size of the cluster of matching anchor points.
Fitting score
The fitting score is defined as 1 - cross correlation score. The cross correlation (CC) score is defined as:
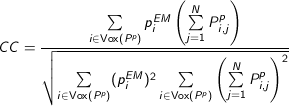 , where
, where 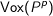 represents all voxels
in the density grid that are within two times the map resolution from any of the atoms of
the protein; and where the total density of P at grid point i is
represents all voxels
in the density grid that are within two times the map resolution from any of the atoms of
the protein; and where the total density of P at grid point i is
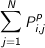 . The values of the CC score range from 0 to 1, where
1 indicates a perfect fit.
. The values of the CC score range from 0 to 1, where
1 indicates a perfect fit.
Dock rotation
The rotation part (in quaternions) of a transformation found by a docking procedure.
Dock translation
The translation part of a transformation found by a docking procedure.
RMSD to reference
If a reference structure is provided (usually in debugging mode), a RMSD to the reference is reported.
Examples
For symmetry example in building a chaperon ring, set the input data as follows:
- Number of Cn Symmetry set to 7
- EM density map

- Resolution is set to 11.5
- Spacing is set to 2.7
- Density threshold is set to 0.852
- X, Y, Z origins are set to -50, -50, -50
- Subunit file
|
| BioInfo3D Servers for protein structure analysis and modeling. |
|
| EMDB Database for electron microscopy density maps. |
E. Tjioe, K. Lasker, B. Webb, H. Wolfson and A. Sali, Nucleic Acids Research, (2011) 39, W167-W170

K. Lasker, M. Topf, A. Sali and H. Wolfson, Journal of Molecular Biology, (2009) 388, 180-194